Birla Institute of Technology (BIT Mesra) 2007 B.E BIOMOLECULAR MODELLING - Question Paper
BIRLA INSTITUTE OF TECHNOLOGY AND SCIENCE, PILANI (RAJ.)
FIRST SEMESTER 2007-2008
BIO C417, BIOMOLECULAR MODELING
COMPREHENSIVE exam
TOTAL WEITAGE 30% Date: 03.12.2007 DURATION: 3Hrs. (Part A & Part B)
Total Marks (70+30) =100
• Answer Part A and Part B in separate ans sheets.
PART – A (CLOSED BOOK) (Max. duration: two Hrs., Max. Marks 70)
1. Write notes on the subsequent
(i) Protein secondary structure (ii) Short-range and long-range contacts in protein folding theories (iii) Protein structure prediction (iv) Hydrogen bonding schemes within DNA bases(v) Simplex method of energy minimization
[4X5=20]
2. Normally when the DNA double helix is right handed the twist and propeller twist are positive to decrease the access of water to the base. For a left handed double helix what would be the sense of propeller twist needed to decrease the access of water to the bases? Schematically show positive and negative propeller twist and twist. [2+8]
3. The table beneath provide the approximate uniform values of roll (R), slide (S) and twist (T) at the base-pair steps in 3 well known forms of DNA “A”, “B” and “C”. Using the subsequent equations compute ‘rise’ tilt, distances of the centers of base-pairs from the axis of the double helix and identify which kind belongs to which form of DNA and justify your ans.
(i) Tilt from axis = (R/2)/sin(T/2) (ii) Length along axis = 3.3 cos(Tilt)+S sin(Tilt)
(iii) Distance from axis= (-S/2) cos(Tilt)/sin(T/2)+3.3sin(Tilt)/2sin(T/2)
DNA kind 1 DNA kind 2 DNA kind 3
R(°) -6 0 +12
S(Å) +1 0 -1.5
T(°) 40 36 32
[6+4]
3. a) What could be the advantages of using SMILE and TOP representation of biomolecules?
b) Does the secondary structure prediction help in the prediction of the tertiary structure of protein? Justify your ans.
c) Comment on the standard energy function in threading technique.
[4+4+2]
4. discuss all steps 1 would follow to perform a molecular dynamics simulation of 10-mer DNA double strand. [10]
5. a) Derive Verlet-leapfrog algorithm to show that the position is determined at every time step whereas velocity is determined at half time-step.
b) What do you mean by sequence dependent DNA structure? What general pattern you observe in sequence dependent DNA structure?
[5+5]
************************************************************************
BIRLA INSTITUTE OF TECHNOLOGY AND SCIENCE, PILANI (RAJ.)
FIRST SEMESTER 2007-2008
BIO C417, BIOMOLECULAR MODELING
COMPREHENSIVE exam
TOTAL WEITAGE 40% Date: 03.12.2007 DURATION: 3Hrs. (Part A & Part B)
Total Marks (70+30) =100
PART – B (OPEN BOOK) (Max. duration: one Hr., Max. Marks 30)
1. In paper electrophoresis at pH 8.6 normal human hemoglobin was run in comparison with 4 abnormal hemoglobin in which amino acid substitution had occurred in the polypeptide chain of 1 kind of subunit. The subsequent outcome was obtained:
Amino acid analysis showed that in 4 hemoglobin the subsequent amino acid substitution had occurred:
A. Lysine was changed by Glutamate in the ?-chains.
B. Glutamate was changed by Lysine in the ?-chains.
C. Glycine was changed by Aspartate in the ?-chains.
D. Asparagine was changed by Lysine in the ?-chains.
(i) discuss which of the hemoglobins A-D corresponds with sample positions1-4 on the electrophoresis strip. [2]
(ii) Would it possible to detect a hemoglobin variant in which glycine in the ?-chain was changed by alanine? Why? [0.5+1]
(iii) Would you expect the charged residues referred to above to be obtained on the surface or in the interior of the protein molecule? discuss. [0.5+1]
Hints:
The subsequent diagram explains the function of paper electrophoresis.
PTO
2. a) How much will it be feasible to model DNA double helix structure through homology modeling? Justify your ans
b) Compare molecular dynamics simulation and Monte Carlo simulation.
c) Write a short review on higher order DNA structure.
[5+5+5]
3. learn the subsequent methodology of molecular dynamics simulation and write down all the assumption made in the provided simulation protocol and comment on the methodology. [10]
Method
We generated tens of thousands of independent trajectories for the 36-residue villin headpiece with molecular dynamics. The bulk of the simulations was performed on a subset of the nearly 200 000 processors around the world participating in our ongoing Folding@Home distributed computing project. We adapted the GROMACS 3.1.4 molecular dynamics package to our distributed infrastructure. Single precision calculation was utilized, as was the case with previously published works with GROMACS and villin specifically, and as with previous works with GROMACS more generally (free energy calculation and protein folding kinetics). We largely followed the setup of Duan and Kollman as regards the temperature and pressure control algorithms, water model, box type, and time step. As in that work, the villin sequence used was MLSDEDFKAVFGMTRSAFANLPLWKQQNLKKEKGLF protein data bank (PDB code 1VII), with N-acetyl and C-amino caps. The protein was solvated for all simulations in 5600–6000 explicit TIP3P water molecules in a truncated octahedron box, with periodic boundary conditions. The minimum distance ranging from a protein atom and the closest image atom was one nm. 3 sodium and 5 chloride ions were included to counter the protein’s charge (30 and 50 mM). Simulations used a two fs integration step and 20 fs neighbor list update frequency, at 300 K temperature. Berendsen temperature and pressure control were used, with coupling time constants of 0.1 and one ps, respectively. Despite its unphysical scaling of kinetic energies, simulations with Berendsen control have previously yielded right folding rates on the microsecond scale. The linear constraint solver (LINCS) algorithm was used to constrain all bonds, rather than only bonds involving hydrogen atoms as in the Kollman simulation. The Garcia-Sanbonmatsu replaced version of AMBER94 (“AMBERGS”) served as the force field. 1 to 4 van der Waals forces were scaled by 0.5 as in the base AMBER94.
Two sets of computations were run, every with a various treatment for long range electrostatics–particle mesh Ewald (PME) or reaction field (RF) Under RF, the Coulombic and van der Waals (vdW) neighbor lists went up to 10 Å with vdW interactions smoothed from eight Å and an external dielectric of 80 was used. Under PME, the neighbor lists went up to eight Å with vdW interactions smoothed from six Å. The grid spacing for Fourier transforms was 1.2 Å, the alpha parameter was 0.39 Å-1, and the interpolation order was 4. The outcomes presented beneath are from the more thoroughly sampled RF ensembles unless otherwise noted.
************************************************************************
BIRLA INSTITUTE OF TECHNOLOGY AND SCIENCE, PILANI (RAJ.)
FIRST SEMESTER 2007-2008
BIO C417, BIOMOLECULAR MODELING
COMPREHENSIVE EXAMINATION
TOTAL WEITAGE 30% Date: 03.12.2007 DURATION: 3Hrs. (Part A & Part B)
![]() Total
Marks (70+30) =100
Total
Marks (70+30) =100
- Answer Part A and Part B in separate answer sheets.
![]()
PART A (CLOSED BOOK) (Max. duration: 2 Hrs., Max. Marks 70)
1. Write notes on the following
(i) Protein secondary structure (ii) Short-range and long-range contacts in protein folding theories (iii) Protein structure prediction (iv) Hydrogen bonding schemes within DNA bases(v) Simplex method of energy minimization
[4X5=20]
2. Normally when the DNA double helix is right handed the twist and propeller twist are positive to reduce the access of water to the base. For a left handed double helix what would be the sense of propeller twist required to reduce the access of water to the bases? Schematically show positive and negative propeller twist and twist. [2+8]
3. The table below gives the approximate uniform values of roll (R), slide (S) and twist (T) at the base-pair steps in three well known forms of DNA A, B and C. Using the following equations calculate rise tilt, distances of the centers of base-pairs from the axis of the double helix and identify which type belongs to which form of DNA and justify your answer.
(i) Tilt from axis = (R/2)/sin(T/2) (ii) Length along axis = 3.3 cos(Tilt)+S sin(Tilt)
(iii) Distance from axis= (-S/2) cos(Tilt)/sin(T/2)+3.3sin(Tilt)/2sin(T/2)
|
|
DNA type 1 |
DNA type 2 |
DNA type 3 |
|
R() |
-6 |
0 |
+12 |
|
S() |
+1 |
0 |
-1.5 |
|
T() |
40 |
36 |
32 |
[6+4]
3. a) What could be the advantages of using SMILE and TOP representation of biomolecules?
b) Does the secondary structure prediction help in the prediction of the tertiary structure of protein? Justify your answer.
c) Comment on the standard energy function in threading technique.
[4+4+2]
4. Explain all steps one would follow to perform a molecular dynamics simulation of 10-mer DNA double strand. [10]
5. a) Derive Verlet-leapfrog algorithm to show that the position is determined at each time step whereas velocity is determined at half time-step.
b) What do you mean by sequence dependent DNA structure? What general trend you observe in sequence dependent DNA structure?
[5+5]
************************************************************************
BIRLA INSTITUTE OF TECHNOLOGY AND SCIENCE, PILANI (RAJ.)
FIRST SEMESTER 2007-2008
BIO C417, BIOMOLECULAR MODELING
COMPREHENSIVE EXAMINATION
TOTAL WEITAGE 40% Date: 03.12.2007 DURATION: 3Hrs. (Part A & Part B)
![]() Total
Marks (70+30) =100
Total
Marks (70+30) =100
PART B (OPEN BOOK) (Max. duration: 1 Hr., Max. Marks 30)
1. In paper electrophoresis at pH 8.6 normal human hemoglobin was run in comparison with four abnormal hemoglobin in which amino acid substitution had occurred in the polypeptide chain of one type of subunit. The following result was obtained:
Amino acid analysis showed that in four hemoglobin the following amino acid substitution had occurred:
A. Lysine was replaced by Glutamate in the b-chains.
B. Glutamate was replaced by Lysine in the b-chains.
C. Glycine was replaced by Aspartate in the b-chains.
D. Asparagine was replaced by Lysine in the b-chains.
(i) Explain which of the hemoglobins A-D corresponds with sample positions1-4 on the electrophoresis strip. [2]
(ii) Would it possible to detect a hemoglobin variant in which glycine in the b-chain was replaced by alanine? Why? [0.5+1]
(iii) Would you expect the charged residues referred to above to be found on the surface or in the interior of the protein molecule? Explain. [0.5+1]
Hints:
The following diagram explains the function of paper electrophoresis.
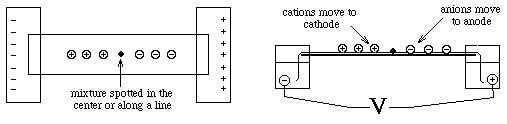
PTO
2. a) How much will it be feasible to model DNA double helix structure through homology modeling? Justify your answer
b) Compare molecular dynamics simulation and Monte Carlo simulation.
c) Write a short review on higher order DNA structure.
[5+5+5]
3. Read the following methodology of molecular dynamics simulation and write down all the assumption made in the given simulation protocol and comment on the methodology. [10]
Method
We generated tens of thousands of independent trajectories for the 36-residue villin headpiece with molecular dynamics. The bulk of the simulations was performed on a subset of the nearly 200 000 processors around the world participating in our ongoing Folding@Home distributed computing project. We adapted the GROMACS 3.1.4 molecular dynamics package to our distributed infrastructure. Single precision computation was utilized, as was the case with previously published works with GROMACS and villin specifically, and as with previous works with GROMACS more generally (free energy computation and protein folding kinetics). We largely followed the setup of Duan and Kollman as regards the temperature and pressure control algorithms, water model, box type, and time step. As in that work, the villin sequence used was MLSDEDFKAVFGMTRSAFANLPLWKQQNLKKEKGLF protein data bank (PDB code 1VII), with N-acetyl and C-amino caps. The protein was solvated for all simulations in 56006000 explicit TIP3P water molecules in a truncated octahedron box, with periodic boundary conditions. The minimum distance between a protein atom and the nearest image atom was 1 nm. Three sodium and five chloride ions were included to counter the proteins charge (30 and 50 mM). Simulations used a 2 fs integration step and 20 fs neighbor list update frequency, at 300 K temperature. Berendsen temperature and pressure control were used, with coupling time constants of 0.1 and 1 ps, respectively. Despite its unphysical scaling of kinetic energies, simulations with Berendsen control have previously yielded accurate folding rates on the microsecond scale. The linear constraint solver (LINCS) algorithm was used to constrain all bonds, rather than only bonds involving hydrogen atoms as in the Kollman simulation. The Garcia-Sanbonmatsu modified version of AMBER94 (AMBERGS) served as the force field. One to four van der Waals forces were scaled by 0.5 as in the base AMBER94.
Two sets of calculations were run, each with a different treatment for long range electrostaticsparticle mesh Ewald (PME) or reaction field (RF) Under RF, the Coulombic and van der Waals (vdW) neighbor lists went up to 10 with vdW interactions smoothed from 8 and an external dielectric of 80 was used. Under PME, the neighbor lists went up to 8 with vdW interactions smoothed from 6 . The grid spacing for Fourier transforms was 1.2 , the alpha parameter was 0.39 −1, and the interpolation order was 4. The results presented below are from the more thoroughly sampled RF ensembles unless otherwise noted.
************************************************************************
| Earning: Approval pending. |